AN INCREASE IN:
Urine porphyrin levels (mostly uroporphyrin)
markedly increased
Fecal porphyrin levels
normal or slightly increased
Plasma porphyrin levels
normal or slightly increased
Contact a Recordati Rare
Diseases Representative
Acute intermittent porphyria (AIP) - one of the hepatic porphyrias - is a rare, inherited genetic condition caused by a partial deficiency of the enzyme porphobilinogen (PBG) deaminase,1 which can disrupt normal heme production in the body.2
Exposure to one or more precipitating factors, such as endogenous steroid hormones or certain prescription drugs, may increase the body's need for heme.3 The partial deficiency of PBG deaminase, the third enzyme in the multi-step heme biosynthetic pathway, can become rate-limiting in the production of heme.4 This can cause neurotoxic porphyrin precursors to accumulate as the body continues to signal for additional heme.4






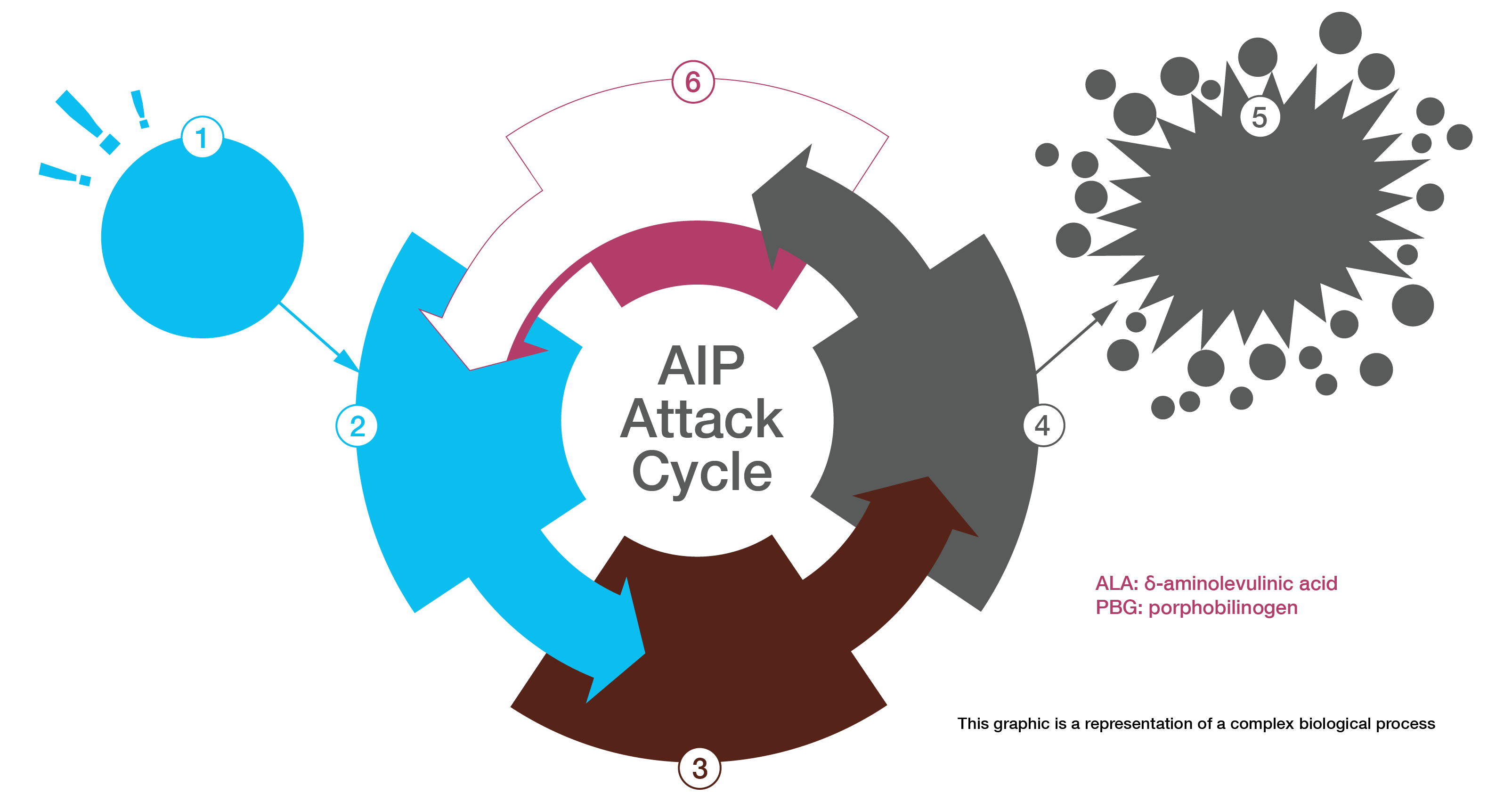
If unrecognized and untreated, an AIP attack can progress to neurologic damage or death due to cardiac arrhythmia or paralysis.3
The most common symptom is severe abdominal pain, which is neuropathic in origin.3 Other symptoms that can increase suspicion of an AIP attack are urine that turns dark or reddish when exposed to light or air and hyponatremia.1 Untreated attacks can cause serious consequences both during and after attacks.3
A PBG urine test should be done at or near the time of AIP symptoms. Urinary PBG levels are substantially increased (20–200 mg/L) in patients with acute attacks of AIP.3
If PBG levels are elevated, second-line testing should be done to determine the precise type of acute porphyria. These tests may include δ-aminolevulinic acid (ALA) and porphyrin testing on the same urine sample, as well as testing for plasma and fecal porphyrins and erythrocyte PBG deaminase. Treatment should not be delayed pending these results.3
can be done to determine the disease-causing mutation(s) in the defective gene. Family members can then be tested to determine if they may be at risk of AIP attacks.3
Urine porphyrin levels (mostly uroporphyrin)
markedly increased
Fecal porphyrin levels
normal or slightly increased
Plasma porphyrin levels
normal or slightly increased
Erythrocyte PBG deaminase levels
decreased by ~50%
PANHEMATIN® is a hemin for injection indicated for the amelioration of recurrent attacks of acute intermittent porphyria temporally related to the menstrual cycle in susceptible women, after initial carbohydrate therapy is known or suspected to be inadequate.
Limitations of Use
You are about to leave the PANHEMATIN.com website and enter a website operated by an independent third party. The links to third-party websites contained on PANHEMATIN.com are provided solely for your convenience. Recordati Rare Diseases does not control the opinions, claims or comments contained on any third-party website linked to PANHEMATIN.com, and your activities at those websites will be governed by the policies and practices of those third parties.